Chapter 8 Genomic Epidemiology of Bacteria
Allison Black
Washington State Department of Health, Seattle, Washington
In our discussion of genomic epidemiology up to this point, we’ve focused on the accumulation of mutations across the genome. While this is common terminology, at this point in the discussion it’s worth clarifying that we have specifically been discussing the accumulation of mutations along the chromosome. For viruses, the chromosome is the only genetic element that they contain. That chromosome can be one or multiple strands of nucleic acid, either RNA or DNA, and may be either single- or double-stranded. The chromosome encodes the genes for making proteins essential for the virus to replicate and assemble functional progeny viruses.
Like viruses, bacteria have chromosomes, but they can carry additional genetic elements as well. Certain genes or entire genetic elements may be present in some but not all bacteria within a species. Still some genetic elements can be present in different bacteria across different species. These patterns of gene gain, loss, and exchange make the bacterial genome complex and dynamic. In this chapter, we’ll break down the different types of genetic loci that contribute to the bacterial genome, and how we tend to use those different genomic elements in genomic epidemiologic analysis. Readers focusing on designing, analysing, or interpreting genomic epidemiological analyses of bacterial diseases should benefit from this chapter.
8.1 Bacterial Mechanisms for Generating Genetic Diversity
Like viruses, bacteria accrue mutations during genome replication. However, unlike RNA viruses whose RNA-dependent RNA polymerases lack proofreading capability, the DNA polymerases that bacteria use to replicate their genomes do have proofreading ability. This means that when the wrong nucleotide is incorporated during genome replication, that error can be fixed. This leads to one of the key differences between viruses and bacteria for genomic epidemiology; bacteria have much slower evolutionary rates compared to viruses. For example, bacterial genomes tend to accrue substitutions at a rate of \(1 \times 10^{- 8}\) to \(1 \times 10^{- 5}\) substitutions per site per year28, while RNA viruses tend to exhibit evolutionary rates between \(1 \times 10^{- 4}\) to \(1 \times 10^{- 3}\) substitutions per site per year10. Even when accounting for differences in genome length, a hypothetical 15,000 nucleotide long viral genome with an evolutionary rate of \(1 \times 10^{- 3}\) substitutions per site per year may accumulate 15 substitutions across the genome in a year, while a five million base pair long bacterial genome with an evolutionary rate of \(1 \times 10^{- 6}\) substitutions per site per year will accumulate five substitutions across the genome in a year.
Since our power to investigate infectious disease epidemiology using genomic data comes from the overlapping timescales of genome evolution and infection transmission, having mutations occur less frequently reduces our ability to resolve transmission, especially on short timescales. Indeed, when a bacterial strain causes an acute outbreak, many if not most of the outbreak-associated cases will have identical or nearly identical core genome sequences. In such cases, genomic analysis sensitively defines which cases form part of the outbreak, but may not provide sufficient information to infer within-outbreak transmission dynamics. For these applications, the primary advantage of whole genome sequence data will be to rule out sporadic cases from an outbreak cluster.
Beyond just their size and their slower evolutionary rates, the practice of bacterial genomic epidemiology is complicated by the fact that sequence diversity is not simply generated through the accumulation of single nucleotide polymorphisms that are inherited by offspring, but also through the loss and acquisition of genetic elements through horizontal gene transfer (HGT) between other bacterial “peers”. We introduce these mechanisms in more detail below; however, one important note is that sequence polymorphisms derived from mechanisms other than clonal evolution should not be used in phylogenetic analyses, as these additional sources of genetic diversity can confound or obscure both the molecular clock and the relationships between isolates. This ability puts the analyst in the tricky situation of needing to determine which SNPs present across the genome occurred through clonal evolution and which occurred though other mechanisms and must be masked from the analysis. While software exists to conduct such decision-making programmatically, the need to know whether the pathogen of interest typically evolves clonally (such as Mycobacterium tuberculosis), or whether diversity might be generated through recombination (such as with many bacterial species including Streptococcus pneumoniae), requires familiarity with your bug, as well as additional effort upstream of your genomic epidemiologic analysis to prepare the dataset.
While we have discussed clonal evolution and the vertical inheritance of mutations in this book already, we will now introduce how genetic diversity can be generated via the uptake of DNA from external sources, whether that be from other somewhat closely related bacteria, different species of bacteria, or the environment, via HGT. Bacteria may inherit small tracts of DNA of a few hundred nucleotides of length, or may inherit large elements encoding multiple genes. Some horizontally acquired genetic sequences integrate into the bacterial chromosome, while others can replicate stably and independently from the chromosome once inside the bacterial cell. Acquisition of this DNA can result in minor or significant genome sequence change if that DNA is integrated into the chromosome, either through homologous or non-homologous recombination. Alternatively, the acquisition of horizontally transferred elements may only change the accessory genome content of the bacterial isolate.
In this section, we introduce these different processes, how they impact genetic variation observed through whole genome sequencing, and how these processes should be used, considered, or corrected for when leveraging bacterial genomic data to support public health investigations.
8.1.1 How Does Horizontally Acquired DNA Get into the Bacterial Cell?
Before horizontally acquired DNA can change the genome of a particular bacterium, it must first be brought into the bacterial cell. There are a myriad of ways in which DNA uptake can occur, with plenty of exceptions and special cases. For the purposes of this text, and this audience, we will not exhaustively review the molecular microbiology of these mechanisms. If the reader is interested though we encourage further study.
There are three primary ways in which a bacterium can uptake external DNA. Firstly, bacteria can take in DNA present in the environment through a process called transformation, in which DNA is brought in through a pore in the bacterial cell membrane. Secondly, in a process called transduction, the genome of a bacteriophage (a virus that infects bacteria, also shortened to phage) is brought into the bacterium upon infection. Some species of phage have genomes that integrate into the bacterial chromosome (lysogenic phage), while other phage species (lytic phage) will simply use the bacterial cell for viral genome replication, package the viral genomes into progeny phage, and kill the bacteria releasing the progeny. The final common mechanism of DNA transfer is conjugation. During conjugation, an apparatus called a pilus acts as a bridge connecting two bacteria directly, facilitating DNA transfer from the donor to the recipient bacterium.
Once inside the cell, what happens to this externally acquired DNA? It may either integrate into the bacterial chromosome, or remain outside of the chromosome. When integration occurs, this single event can lead to either minor or major changes in the nucleotide sequence, depending on the processes governing that integration event. We’ll summarise those processes at a high level in the following sections.
8.1.2 Integration of Horizontally Acquired DNA into the Chromosome
Horizontally acquired DNA often integrates into the chromosome via homologous recombination, a process by which a typically short portion of a bacterium’s genome is replaced by a sequence from a different bacterial lineage when the incoming DNA is similar (homologous) to the chromosomal DNA.
Homologous recombination is an important and fundamental mechanism for bacteria to generate genomic diversity; it is every bit as much a record of evolutionary history as the accumulation of substitutions that arise due to error-prone replication. However, it is a much harder evolutionary signal for us to interpret from an epidemiological perspective, and genomic epidemiologists typically discuss it as an obstacle that must be overcome rather than a signal to investigate. We’ll clarify why this is, and how we commonly approach dealing with this natural process.
As we’ve discussed, clonal evolution is the process by which mutations occur during error-prone genomic replication, are integrated into the sequence, and inherited by descendent organisms. Clonal evolution typically occurs at a steady rate (the molecular clock), and introduces these mutations relatively uniformly across the genome. In contrast, during the process of homologous recombination, a small fragment of similar, but not identical, sequence replaces the homologous area of the genome that was previously present. This event has the effect of introducing multiple changes to the genome sequence during a single event, essentially accelerating the incorporation of polymorphic sites in the genome compared to the frequency that those substitutions would have arisen through clonal evolution alone.
The incorporation of these multiple substitutions in a single event isn’t the sole problem; indeed, if these recombination events occurred a regular intervals and introduced similar numbers of genetic changes per recombination event, we’d recapture a steady rate at which genome diversity accumulates, albeit through two processes (recombination and clonal evolution). We are not that lucky. Homologous recombination appears to occur with irregular frequency, and the number of genetic changes, or even additions or deletions of whole genes, that occur during a single recombination event is highly variable. Furthermore, the length of DNA incorporated into the genome (referred to as the “track length”) can vary tremendously, from hundreds to tens of thousands of base pairs. These factors thereby impact the correlation between the passage of time and the acquisition of substitutions across the genome, undermining our ability to accurately estimate evolutionary rates.
If the challenges imparted to estimating the molecular clock weren’t tricky enough, homologous recombination, by the very fact that it introduces a different piece of genome into the recipient bacteria, creates a situation where different parts of the genome have different evolutionary histories. Sequences introduced through homologous recombination can make a genome look more diverged from other sequenced samples in an analysis or make sequences look less diverged, and more closely related, than they really are. In essence, different portions of the genome will be more or less related to other sequences – the evolutionary history written into the genome is different depending on which portion of the genome you’re looking at. This reality undermines our ability to infer clear and consistent relationships between analysed samples, and the phylogenetic trees which are usually the object that we use to describe those relationships.
These challenges explain why we often treat recombination as a nuisance to weed out rather than an evolutionary process to study. So how do we handle the presence of recombination? Typically, we seek to remove sequence polymorphisms that we believe are attributed to recombination, leaving us to analyse only those substitutions in the genome that we believe occurred due to clonal evolution. Typically, we use statistical methods to detect recombinant portions of the genome characterised by a high density of substitutions within a limited sequence fragment length. Those areas will be masked, leaving us the vertically inherited SNPs to use in our phylogenetic analyses.
Importantly, this method of detecting recombination really only works with closely-related sequences, where the receipt and integration of a new genomic fragment results in an apparent abundance of mutations that is easily distinguished from the frequency of mutations in surrounding areas of the genome. When analyzing highly diverged sequences (e.g., a global dataset of a particular bacterial species), sequences will hold evidence of recombination receipt and donation, and recombination can make sequences look more divergent or more similar to each other than would be apparent from clonal evolution alone. This makes inferring the deeper historical relationships between bacteria exceptionally difficult. However, from the applied genomic epidemiology perspective, we’re fortunate to generally investigate infection transmission on the timescale of days and months. The genomic diversity of an outbreak-attributable strain is typically very low within the outbreak, and recombination detection is possible.
8.1.3 Acquisition and Exchange of Extrachromosomal DNA
Thus far we’ve discussed how integrating externally acquired DNA into the bacterial chromosome can introduce new polymorphisms to the genome. However what happens when that externally acquired DNA does not integrate into the chromosome? Here, we’ll discuss the impact of plasmid transmission on gene content and function, with a particular focus on public health impacts and genomic analysis.
8.1.3.1 Plasmids
Plasmids are sufficiently important from a public health perspective that we will discuss them in detail. Plasmids are extrachromosomal DNA elements, usually, but not always, made up of circular double-stranded DNA, and they harbour genes encoding proteins that might provide benefit to the bacterium in certain environments. They do not encode the most essential genes that the bacterium needs for replication. Rather, you can think of plasmids as carrying “bonus material” for the bacterium. Plasmids can range in size from 2 to 5 kilobase pairs (Kbps) to up to 500 Kbps.
Most commonly, plasmids replicate independently of the chromosome, although some plasmids can integrate and excise themselves from the chromosome, and when integrated get replicated during the process of chromosomal replication. Bacteria can gain and lose plasmids, and they can harbour more than one plasmid, although certain plasmids may not be able to stably co-exist within a bacterium, a feature known as plasmid incompatibility. Furthermore, plasmids can be transmitted between different bacteria, both within and between bacterial species, via HGT.
8.1.3.2 Analysis
Depending on the sequencing and genome assembly approach (discussed in more detail later in this chapter), plasmids found in bacterial strains can be partially or fully assembled. Oftentimes, review of the annotation files will show phage-associated coding sequences, and depending on the species, plasmid “typing” tools exist to identify well-known plasmids. This identification is usually made based on the gene content of the plasmids. When carrying out analysis on a large sample of bacterial genomes, one common component is to identify the presence and absence of plasmids or at least the epidemiologically important genes such as antimicrobial resistance determinants, that are found on them. This can be particularly important when investigating plasmid-mediated outbreaks. For these investigations, tracking the plasmid may be the primary focus of the investigation, even in instances where the bacteria harbouring them are different.
8.1.3.3 Limitations
Plasmid assembly can be hard using short read sequence data because portions of the plasmid may have sequence homology with the chromosome or even contain repeat regions within the plasmid. In addition, plasmids are often present in high copy numbers in relation to the bacterial chromosome. Together, this complicates the assembly process, making it hard to finish plasmid sequences. Long read sequencing and hybrid assembly, discussed in Section 8.5.1, can sometimes be used to obtain complete plasmid assemblies.
8.2 A Brief Note About Bacterial Population Structure
Several chapters could be spent discussing how and when bacteria get separated into subpopulations that evolve distinctly from each other (population structure) and bacterial speciation. Simply, the population structure of bacterial species is complex. As described, genetic variation in bacteria is driven by recombination and mutation, and that variation is acted upon by selection and chance. Combined with other biological and ecological factors, these processes can shape the structure of a species or blur the boundaries between them. This process is ongoing and large changes usually occur over longer evolutionary timescales.
Thankfully, outbreak investigations mostly focus on a subpopulation of a bacterial species over shorter timescales. How structured a bacterial population is ranges from completely unstructured (termed panmictic) to well-structured (or clonal). Where a species falls on this spectrum is measured by how much statistical association is found among loci on the genome, referred to as linkage disequilibrium. Without diving into a detailed discussion on bacterial population genetics, the important concept is that the more recombination a species experiences, the more mixed it becomes, and the less structured the population29. This of course is an oversimplification. In practice, few bacterial species are regarded as truly panmictic; Helicobacter pylori and some environmental species of Vibrio are included in that group. Staphylococcus aureus and Mycobacterium tuberculosis are canonically referred to as clonal, while most others, such as species of Neisseria and Streptococcus, fall in the middle. When investigating a putative outbreak, it is important to have a general knowledge of the suspected species’ population structure.
8.3 Bacterial Sequence Typing
Prior to the advent of whole genome sequencing, many serological and molecular based typing methods were developed to define bacterial population structure and classify strains, several of which are species specific. These include multi-locus sequence typing (MLST), pulsed-field gel electrophoresis (PFGE), spa typing for S. aureus, restriction fragment length polymorphism (RFLP) analysis, serogrouping/serotyping, and others. The most common, MLST, uses allele profiling in seven housekeeping genes to define the sequence type of a particular bacterial strain. Each bacterial species has a specific MLST scheme, which can be found at https://pubmlst.org/. For each MLST scheme, the particular nucleotide sequence of each gene is summarised with a label, which is typically a number. When another strain has the exact same nucleotide sequence at that same loci, they are annotated with the same number. If a particular strain has a sequence never before observed at a particular loci, then that sequence will be provided a new numerical label. This allows a bacterial strain to be summarised according to their sequence type (ST) – whereby the numerical labels that summarise each loci map back to a particular sequence motif, and make comparing sequence identity across a large genome more wieldy.
The trade-off for this easy annotation is that when your strains have different sequence types, the MLST scheme will not summarise the amount of genetic distance between your different sequence types. The MLST annotation provides a singular label to define a sequence motif across particular genetic loci. When you observe two different labels for the same loci, that information does not tell you whether those two sequence motifs differ by one or tens or hundreds of substitutions. To complicate things, a single mutation in a housekeeping gene may result in the assignment of a new ST. In addition, the same ST can wind up on different genomic backgrounds through recombination, and the same STs may even de novo evolve in different lineages. As with many things, the ease that comes from MLST also abstracts away some of the data richness, which you may find useful to return to when diving into bacterial genome analysis. With greater access to cost-efficient sequencing, we can now sequence larger portions of the genome, leading to the ability to define a multi-locus sequence type using all genes present in the core genome. This type of MLST, often referred to as cgMLST – core genome multi-locus sequence typing – recently rose in popularity especially for use in large genomic databases. However, for conducting genomic epidemiology studies, all variation found in the “core genome” (detailed below) is routinely used for analysis.
8.4 Defining Bacterial Genomic Elements
Typically, we discuss bacterial genome organization in terms of the core genome, the accessory genome, or the pangenome.
8.4.1 Core Genome
The core genome is defined as the set of genes that are present across all strains within a particular sample from a bacterial species. This is a functional definition as the core genome changes based on the samples you are looking at, which could include members of a single bacterial lineage, species, or even several closely related species. As genome content varies considerably even among members of the same species, the core genome, by definition, is that portion of the genome that is present and comparable across all samples of interest.
8.4.1.1 Analysis
Genomic epidemiological analysis of the core genome typically focuses on defining relationships between isolates according to the patterns of shared and unique substitutions, acquired through error-prone replication, observed across different isolates’ core genomes. With core genome analysis we regain much of the ease of viral genomic epidemiology. Namely, by using the core genome we ensure that we’re looking at genomic regions where we have information for all samples; we do not have to consider how to incorporate gene presence/absence information. And by focusing on single nucleotide polymorphisms (SNPs) accrued across the core genome through clonal evolution, we can approach genomic epidemiological analysis using the same principles as we’ve discussed for viruses. We can align sequences to each other, compare which sites in that alignment show sequence polymorphism, and use that information to perform phylogenetic analysis.
Since bacteria have much larger genomes than viruses, they can be harder to work with computationally. This situation means that while for viral genomic epidemiology we tend to store, share, and analyse the entire genome sequence, for bacteria we will often consolidate and summarise sequence identity, either by only looking at portions of a multiple sequence alignment where sequence polymorphisms exist, or by summarizing sequence identity with a categorical annotation (e.g., MLST, as defined previously).
The size of the core genome will decrease as the number of samples you are considering increases. The reason for this is that as the diversity – both in terms of genome content and nucleotide variation – of the sample set increases, fewer genes are shared across all samples in the set. An extreme example of this phenomenon would be if you were to analyse the core genome of samples from different bacterial species. The set of genes present across multiple bacterial species will be much smaller than the shared set of genes present within a particular clade of a single species. From a phylogenetic interpretation perspective, this is why dominant clades in a species-level phylogeny will often have very short terminal branch lengths. With a core genome that has been whittled down to genes present across multiple species, there’s much less sequence (and therefore genetic variation) observable within the species or clade.
8.4.2 Accessory Genome
The accessory genome is all of the genetic elements, either on the chromosome or harboured on mobile genetic elements such as plasmids, phages, or integrative and conjugative elements, that are present within a particular bacterial strain, but are not found across all samples in your sample set. These genes are often epidemiologically relevant, conferring antibiotic resistance or virulence (such as biofilm formation or toxin production), and can come and go depending on availability and the needs of the organism in a particular environment. In some instances, “core genes” impacted by high rates of mutation and/or recombination can be so diverged that they cluster separately. In these cases, such as for S. pneumoniae genes pspA and pspC, a “variant” of that gene can be found in every member of the species, but the amount of diversity precludes the ability to align all of them. Therefore, they wind up in the accessory genome. Again, this emphasises that this system of categorization is largely functional rather than biological.
8.4.2.1 Analysis
For bacteria with dynamic genomes, meaning that they experience frequent gene acquisition and gene loss, looking at gene content variation across the accessory genome can provide an additional source of information when differentiating between cases. For example, during a hospital-associated outbreak of Acinetobacter baumannii, Mateo-Estrada and colleagues30 found that many cases had identical core genome sequences, even though cases and sample collection had occurred over multiple years. However, when applying a simple assessment of whether a gene was present or absent from a sample’s accessory genome, they were able to capture genomic content divergence between the different outbreak cases.
8.4.2.2 Limitations
While genome content variation provides additional resolution to observe genetic similarity or dissimilarity between cases, it may be challenging to use that data beyond a determination that cases are truly clonal (identical) or not. Specifically, it’s much harder to use gene content variation to investigate transmission over time, which is how we’ve typically discussed evolutionary analysis throughout this book. When changes to the genome occur by chance and are inherited by descendents through clonal evolution, the degree of sequence identity observed parallels the amount of transmission that separates cases. However, when genes can be easily lost or acquired, and this process may be driven by selective pressures or the environment, it becomes much harder to assess what it means epidemiologically to share a genetic element. Specifically, do two cases share a particular accessory gene because they have epidemiologically linked infections, and one case inherited that genome organization from the primary infection? Or are the cases epidemiologically unrelated, but both occurred in a similar ecological setting – such as the use of a particular antibiotic – where the acquisition of an antimicrobial resistance gene would confer an advantage? Finally, as with all computational analysis, errors in genome assembly and annotation can result in spurious differences in gene content.
8.4.3 Pangenome
The pangenome represents all genetic elements present in a bacterial isolate – both the core genome and the accessory genome together. When genomic epidemiologic studies consider both phylogenetic patterns implied from core genome SNP data and patterns of variation in accessory genome content as information about organism relatedness, we term such analyses “pangenomic epidemiology”. A notable characteristic of pangenome variation is that, due to patterns of recombination, core genome SNP diversity and accessory genome diversity (measured by the presence and absence of accessory genes) are correlated. The more distantly related members of a species are in their core genome SNPs, the more dissimilar their accessory genomes. This is somewhat intuitive if we consider population structure, as briefly described above.
8.5 Bacterial Genome Sequencing and Assembly
To generate whole genome sequences for viral genomic epidemiological analysis, the lab will often employ amplicon-based sequencing followed by mapping of sequencing reads to a closely related reference genome, facilitating the assembly of those reads into a consensus genome. For bacteria, with their genome content variation and extensive within-species diversity, there are some nuances to genome assembly and analysis that should be considered. Below we will describe the main approaches for bacterial genome assembly, and how each approach may impact downstream inference of relatedness.
The two main approaches to bacterial genome assembly are reference-based assembly and de novo assembly. For reference-based assembly, sequencing reads are mapped to a closely related reference genome, mirroring the amplicon-based approach we commonly use for viruses. De novo assembly, also referred to as reference-free assembly, uses information from overlapping portions of the sequencing reads to piece the genome together like a jigsaw puzzle. While reference-based assembly will result in a consensus sequence with the same length as the reference genome, de novo assembly will result in several (anywhere from tens to hundreds) of contigs – a contiguous sequence derived from multiple overlapping sequencing reads and representing a large portion of the bacterial genome. Contigs inferred during de novo assembly are then annotated using tools like Prokka31 or NCBI’s Prokaryotic Genome Annotation Pipeline (PGAP)32. Each approach has associated advantages and limitations, which we will discuss, and in practice, the two are often used in tandem.
The main disadvantage of reference-based assembly is that reference selection can significantly impact downstream analysis. Most notably, you can only identify variation in regions of the genome that are shared between the reference and the sequenced bacterial isolates. If the strain of interest possesses mobile genetic elements or genes acquired through HGT that are not present in the reference, then the ability to identify SNPs in those loci is lost. Overall, the more diverged the reference, the less “callable” the genome. If the reference is diverged to the point that there is an appreciable amount of mutations, insertions and deletions (often shortened as indels), or genome rearrangements, then read mapping could be adversely affected. This could result in an artificial reduction in SNP distances amongst your bacterial isolates of interest, which would likely impact the inference of epidemiological linkage. A good metric to assess the appropriateness of the reference is to measure the percentage of reads mapping to the reference during the mapping step of genome assembly. If the reference is a close match, you should expect to see 96%–99% of sequencing reads mapping to the reference.
With de novo assembly, the organization of the genome is lost, that is, the contigs are not ordered in the way they are found in the bacterial chromosome. As a result, contigs from separate isolates cannot simply be aligned to each other to identify SNPs. The solution to this issue is to first identify all of the coding sequences in the de novo genome assembly through the process of genome annotation. Afterwards, the annotated sequences are used as the input for pangenome analysis programs that then identify and cluster all of the orthologous coding sequences; these are referred to as clusters of orthologous groups or COGs. Subsequently, each COG is individually aligned, which is a far easier computational feat than attempting to align complete bacterial genomes. Afterwards, the core and accessory genome of a set of sequences is defined based on the presence and absence of COGs in the dataset. The alignments for COGs comprising the core genome can then be concatenated into a core genome alignment. It’s worth noting that these alignments do not include intergenic regions of the bacterial chromosome, which contain the biologically relevant gene promoters. In addition, information about the distance between COGs is lost. Further, while core genome alignments only contain a portion of the total genome content, file sizes can still be very large, especially for datasets including hundreds of isolates. To address this, users often extract only the variable sites, creating a core genome SNP alignment. The SNP alignment is then used for downstream phylogenetic analysis.
8.5.1 Considerations for Long-Read Sequencing Data
Sequencing platforms can broadly be categorised into those that produce “short” and “long” reads. Short read platforms like those from Illumina produce reads ranging from 50 to 350 base pairs while long read sequencers such as those from PacBio and Oxford Nanopore Technology (ONT) generate reads from 1,000 to longer than 10,000 base pairs in length. A key difference is that while short read sequencers produce uniform read lengths, long read sequencers result in a distribution of read lengths. In addition, long read sequencers have lower per-base read accuracy than short read sequencers. Notwithstanding, the popularity of long-read sequencers has grown significantly as platforms like those from ONT offer flexibility in sequencing throughput. For example, there is no set time for the duration of an ONT sequencing run – one can run it until sufficient data has been generated and the flow cell can be reused as long as there are remaining active pores. In addition, base calling occurs in real time, which offers a number of potential applications for diagnostics and genomic epidemiology. An attractive feature of long reads is that they can be used to more accurately determine genome organization. However, people often cite the lower accuracy as a downside of the technology. To address the lower accuracy compared to short reads, methods have been developed to combine short and long read data into hybrid genome assemblies. For hybrid assembly, a short-read-only de novo assembly is first generated, then the long reads are used to order (scaffold) the contigs. The short reads are further used to correct any errors in the assembly by iteratively mapping the reads to the draft genome in a process called “polishing”. It is not uncommon for this approach to result in a fully circularised “closed” genome. While the ability of long read sequencing data to create closed genomes is certainly impressive, at present, it likely provides a negligible advantage for most goals of genomic epidemiology analysis. As the accuracy of long read sequencers improves, long-read only de novo assemblies may present an alternative for regular use. At the time of writing, ONT had released updated flow cells and corresponding library preparation chemistry that were capable of \(> 99\%\) accuracy.
8.6 A Practical Workflow For Bacterial Genomic Epidemiology
Here, we will provide an overview of a generic workflow for bacterial genomic epidemiology. While the steps may vary based on the bacterial species of interest, the general process is overall consistent. We urge the user to first investigate the genomics of the microbe. This should include an understanding of the basic genome structure (e.g., number of chromosomes, genome length, presence of plasmids, average number of coding sequences, and GC content), whether the species is known to be naturally competent (i.e., able to undergo homologous recombination), and the basics of population structure and molecular typing. We will assume that short-read sequencing data has been generated for a set of isolates, as this is currently the most common platform in public health labs, and that the necessary quality control steps have been performed.
The workflow usually begins with a de novo assembly of all the samples followed by genome annotation. A quality control step at this point involves assessing the statistics from the annotation process to identify the number of assembled contigs, total genome length, GC content, and the number of coding sequences (CDSs) annotated. By plotting the statistics, for example using a basic histogram, the user can flag outliers, which should either be further examined or excluded from the subsequent analysis. De novo assemblies can then be used as inputs for a number of tools to confirm the species, identify genotypic antibiotic resistance and virulence determinants, assign a sequence type, and genotypically assign serotypes or serogroups, if relevant to the species of interest.
After this step, the annotation files are used by pangenome analysis tools to determine the core genome. As detailed above, this process will result in a core-genome SNP alignment that can be used to produce a SNP distance matrix or phylogeny. As a quick technical note about best practices, users should use phylogenetic algorithms that take into account SNP alignments. These algorithms usually include some form of ascertainment bias correction that corrects for the “missing” nucleotides in the alignment that were removed because they did not show polymorphism. In addition, users should be reminded that if they are working with a recombining species, this alignment will also include SNPs introduced through recombination, unless recombination detection and masking was previously performed.
At this point in the analysis, the phylogeny should be visualised in conjunction with any typing, antibiotic resistance, or epidemiological data that are relevant. A common practice is to annotate the phylogeny with the typing information (e.g., MLST) and to use a heatmap to illustrate the epidemiological data associated with each sample. If the goal of the analysis is to identify samples that may be related through recent transmission, then this step can be used to identify samples that can be excluded (i.e., “ruled-out”) from the putative transmission clusters. This will often focus the analysis on a subset of samples. Since the inclusion of “outliers” reduces the core genome size, the terminal branch lengths on the lineage or clade of interest will be artificially lower, as discussed above. To address this, the samples excluded from the putative outbreak cluster, should be removed from the dataset and the pangenome analysis repeated. This secondary analysis will yield more accurate SNP distances that can be used to infer transmission events.
The workflow above avoided the use of reference-based genome assembly and can be used for a variety of applications. However, if the species is known to experience homologous recombination, then the removal of genomic regions that have been impacted by recent recombination events may be necessary. If this is the case, then the above workflow should still be used to identify the clade or cluster of interest. An appropriate reference genome can then be identified using a number of available tools. Thereafter, the sequence reads from each sample in the subset of interest can be mapped to the reference and a full length multiple sequence alignment can be obtained. Thankfully, over the last decade, bioinformatics tools have significantly advanced and readily available tools like Torsten Seemann’s Snippy (https://github.com/tseemann/snippy) can perform much of this analysis. Once created, the multiple sequence alignment will be used as an input for recombination detection tools. The reference based assembled and full length alignment is needed because most recombination analysis tools use the distance between loci to detect evidence for recombination. As noted, these programs will “censor” areas of the genome that have been impacted by recombination and produce a “recombination free” alignment. In addition, they will output several statistics including the ratio of SNPs introduced through recombination compared to mutation (r/m).
8.7 Intrahost Diversity And Implications For Inferring Transmission
As we move towards a discussion about how to infer the relatedness among bacterial isolates using whole genome sequencing data, we must first consider an important aspect of microbial transmission and evolution. A bacterial population present during an episode of asymptomatic carriage or disease is just that, a population.
While most genomic epidemiology studies employ the sequencing of a single isolate, this practice is largely the result of the dogma of classical microbiology techniques combined with computational and cost constraints. Recent studies have described the amount of microbial diversity that can exist in an individual, referred to as intra-host or within-host diversity, which is not captured by sampling only a single isolate. This topic won’t be explored in its entirety here, but a brief overview is needed to understand how intrahost diversity may impact the investigation of transmission dynamics during an outbreak.
When a bacterium is acquired, it begins to replicate. As described previously, uncorrected errors occurring during replication can result in the ongoing accrual of mutations and indels. In addition, bacteria often come into contact with other microbial species and sometimes other members of the same species. This provides the opportunity for acquisition of mobile genetic elements such as phages or plasmids, or in the case of naturally competent bacteria, to undergo homologous recombination using DNA taken up from the environment. The combined effects of these processes results in appreciable diversity within the bacterial population. This diversity may extend beyond nucleotide diversity and include gene content variation.
The amount of diversity that accrues is heavily dependent on the bacterial species and whether the acquisition results in carriage or disease. For carried bacteria, the duration and site of carriage can also impact the breadth of diversity. Sites may include the lower or upper respiratory tract, skin, or gastrointestinal tract, and carriage at multiple anatomic sites is not uncommon. The longer a bacterial population is carried, the more opportunity there is to come in contact with other bacteria and to also accumulate mutations. Put simply, increasing carriage duration results in a larger and a more diverse population. For bacterial acquisitions that result in infection, there is often less opportunity to accumulate diversity since this process usually occurs over shorter timescales.
One potential outcome of acquiring a pathogen is subsequently transmitting that pathogen on. During a transmission event, only a subsample of the total diversity present in a host is transferred. The amount of diversity transferred is referred to as the transmission bottleneck size and is dependent on the mode of transmission and the amount of diversity that was present on the primary host. For example, transmission of a respiratory pathogen via a fomite may have a smaller transmission bottleneck size than direct respiratory droplet transmission. Each transmission event can transfer a different random subsample of diversity. Because of this, sampling a single bacterial isolate from each individual in a transmission chain that involves one person transmitting to multiple recipients will more than likely result in difficulty inferring the direction of transmission. This is a current limitation in methodological approaches to inferring transmission. Below we will discuss potential future solutions. For a more complete discussion of within host evolution, Didelot and colleagues33 provide an excellent review.
8.8 Investigation of Transmission
The application of bacterial whole genome sequencing to investigating transmission has provided an unprecedented level of resolution beyond what was previously capable with traditional molecular typing approaches. Still, the process of inferring transmission using genomic data remains complex and is often debated. The approaches, ordered by increasing complexity, are 1) SNP-distance based methods, 2) phylogenetic methods, and 3) modeling based methods. Before diving into the details of each, the one constant across all approaches is that epidemiological data must be taken into consideration when inferring transmission. No pathogen-centric method, sequencing or otherwise, will ever replace the need for a classic shoe-leather epidemiology investigation. Another truism is that it is far easier to rule-out a case using pathogen genomic data than it is to rule-in.
SNP-distance based methods are the easiest and most simplistic method for ruling-in and -out cases from a putative transmission cluster. This method uses a SNP cutoff to define isolates from cases likely related by recent transmission. These cutoffs can be set a priori based on previous outbreak investigations and knowledge of the organism or empirically based on the data collected during the investigation at hand. For example, SNP cutoffs for S. aureus have long been held as ranging from 30 to 40 SNPs between related isolates. An extensive study of S. aureus transmission modeling later found that a cutoff of 39 SNPs34 yielded a high sensitivity and specificity for inferring transmission. Of course, since genome size, recombination rates, and evolutionary rates vary greatly among bacterial species, one single cutoff is not suitable for all.
If an analyst obtains a general sample of bacterial genomes from a species of interest as well as those that are suspected as being from an outbreak, then they can empirically define a SNP cutoff. To do this, one obtains a pairwise SNP distance matrix and then plots the distribution of SNP distances using a histogram (see Figure 8.1 for an example). This distribution will be bi- or tri-modal (having two to three peaks). The furthest right peak will illustrate the SNP distances between distantly related members of the species, the middle peak will represent the members of a lineage or clone of that species, and the furthest left peak will be the genomes that are the most closely related – and most likely to be linked by recent transmission. Therefore, you could set your SNP cutoff to fall between the first and second peak (looking from left to right). When combined with epidemiological data (e.g., overlapping lengths of stay in a hospital, consumption of the same food item, or sexual contact), this may be all you need to resolve the outbreak. Increasingly, methods such as those from Duval and colleagues35 facilitate the estimation of outbreak specific SNP thresholds, accounting for organism and sample-specific evolutionary rates, and variation in outbreak duration.
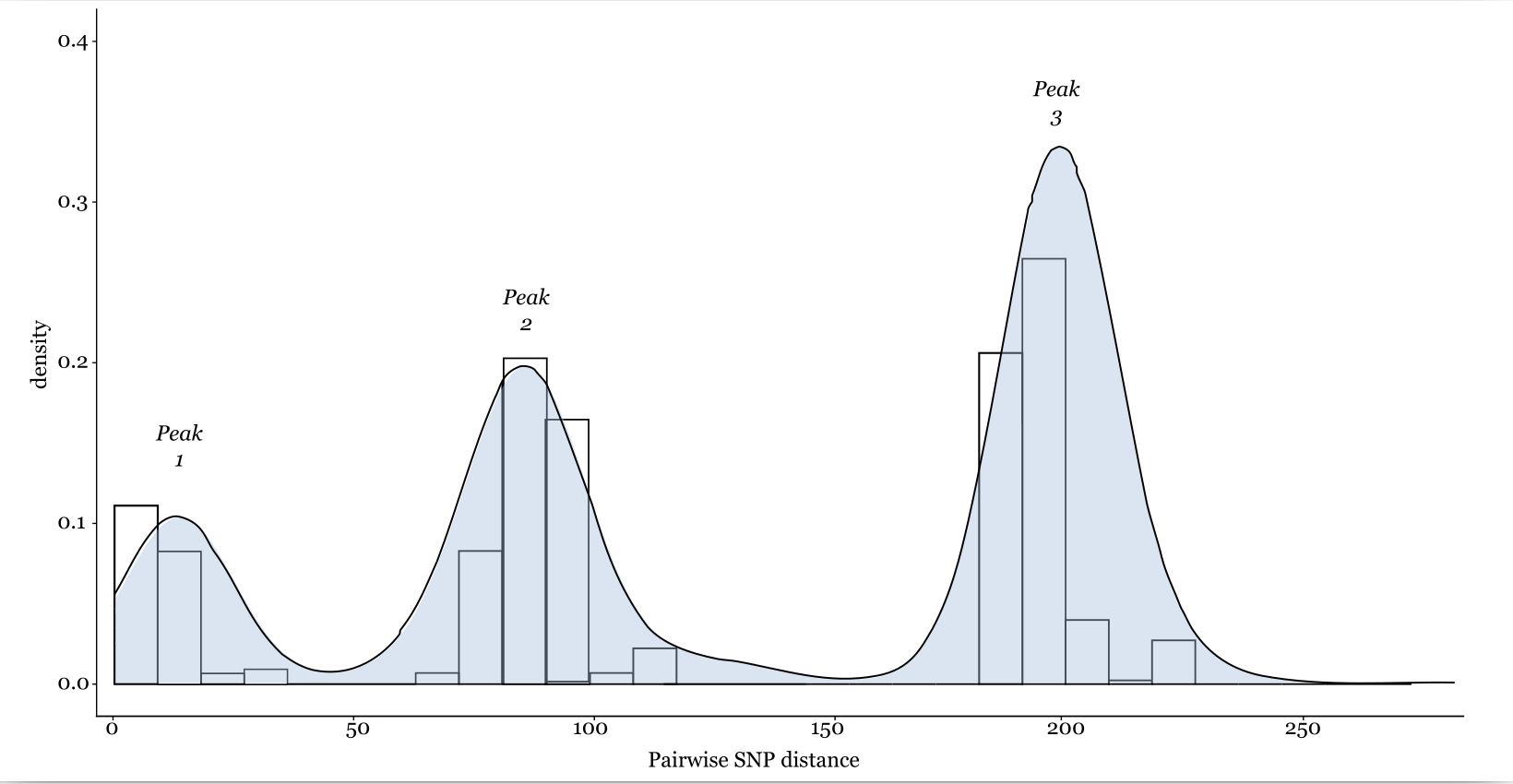
Figure 8.1: A histogram of theoretical SNP distances between various bacterial core genome sequences. In this case, the distribution is trimodal. A small number of sequences cluster together with very few SNP differences, likely signalling relatedness, while other groups of isolates are somewhat more different from each other.
Phylogenetic methods are more complex and not as easy to interpret, a classical example of increased data richness also yielding greater nuance and clearer underlying uncertainty. A staunch phylogeneticist would argue that you are “throwing away” important data by only considering the number of SNP differences and not incorporating our understanding of molecular evolution. Therefore, one should use a phylogenetic tree inferred from the core genome alignment to investigate transmission. The catch is that one’s interpretation of the tree is always caveated since the phylogenetic tree does not always match the transmission tree. In addition, evolutionary distances used when displaying a tree may not be readily interpretable for analysts new to reading trees. Nevertheless, phylogenetic-based inferences of transmission are likely to provide the best resolution when inferring transmission patterns. As an extension of the current phylogenetic approaches, methods are being developed to incorporate sequencing from high density sampling (i.e., sequencing up to eight to ten isolates from a single case) or high depth sequencing of the entire bacterial population, referred to as deep sequencing. These approaches address the issue of intrahost diversity and promise the ability to infer the directionality of transmission36. The goal here is to identify whether the “cloud of diversity” characterizing one case’s bacterial population is nested within the genetic diversity of another individual’s bacterial population. The more the diversity overlaps, the more confidence there is that the populations are directly related through transmission37. When a temporal component is incorporated into the analysis, then the directionality of transmission can be inferred.
The term “modeling based methods” is used here to describe computational approaches that incorporate epidemiological and pathogen genomic data to jointly infer transmission, such as TransPhylo39. Epidemiological data could include dates of disease onset and laboratory report dates as well as estimates of the infectious period. Values such as the transmission bottleneck size can also be estimated. An advantage of this approach is that it provides a probability of transmission between two cases. This is particularly appealing because public health decision makers often want to know how confident analysts are in their findings when a simple “yes/no” or “rule-in/out” answer is not possible (which it rarely ever is). The disadvantage is that these methods are largely accessible to only advanced analysts. However, as these methods are improved upon, they could eventually be incorporated into popular genomic epidemiology graphical user interfaces. In addition, as sampling strategies evolve, the use of deep sequencing or high-density sampling could significantly advance our ability to determine the true transmission tree34.
8.9 Conclusions
Taken together, due to the complexity of their genomes and unique evolutionary dynamics, the application of genomic epidemiology to bacterial species requires specialised knowledge distinct from viruses. However, much of the methodological approaches are consistent when investigating suspected viral or bacterial outbreaks. There are countless success stories in bacterial genomic epidemiology, and as a result, genomic surveillance of select bacterial pathogens is routine in many jurisdictions. Large bacterial genomic databases are now available for several species, which can be used to inform our knowledge about diversity and distribution of the population. Again, one must consider the nuances of bacterial evolution when investigating transmission. This includes an understanding of 1) the epidemiology of the pathogen, 2) recombination and mutation and how they shape population structure with the help of selection and drift, 3) genome content variation and pangenome analysis, and 4) intrahost evolution. With ever-evolving computational tools, bacterial genomic epidemiology is becoming more and more commonplace. The hope is that this will ultimately lead to earlier identification of emerging threats and rapid case investigation to mitigate spread.