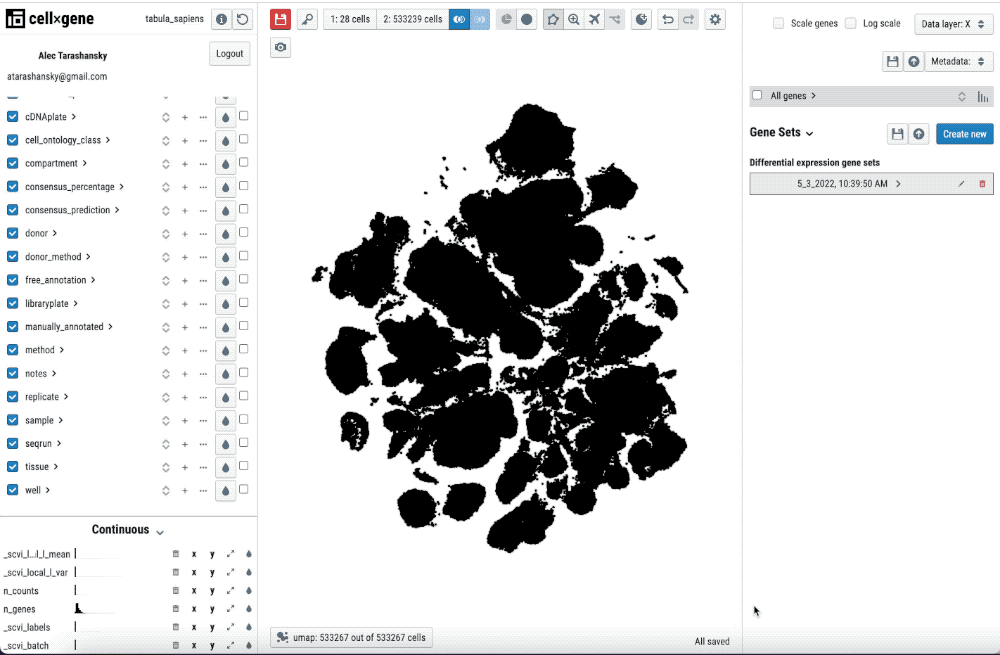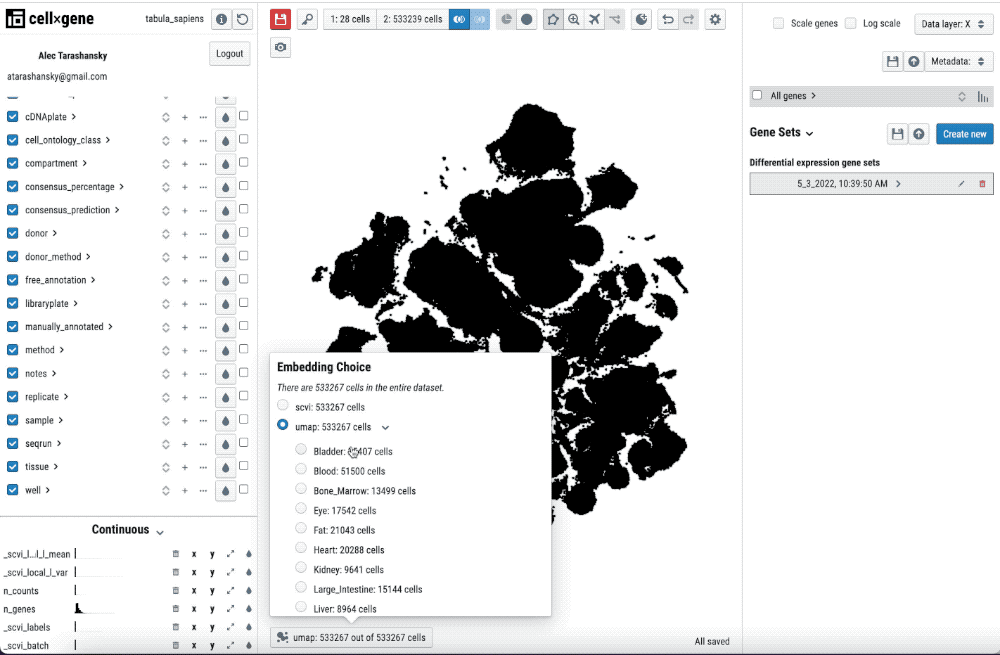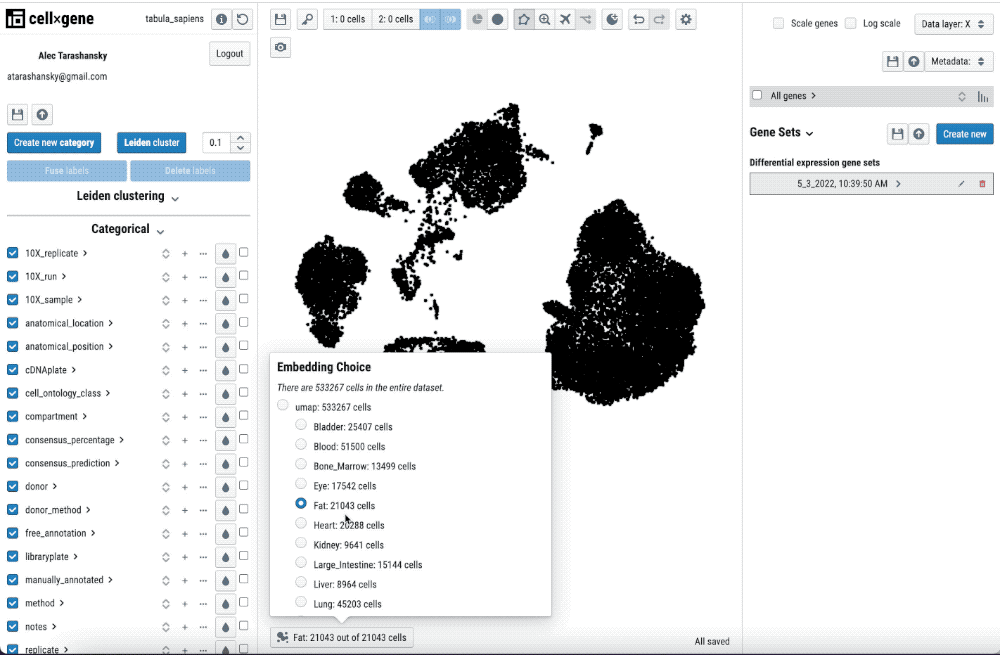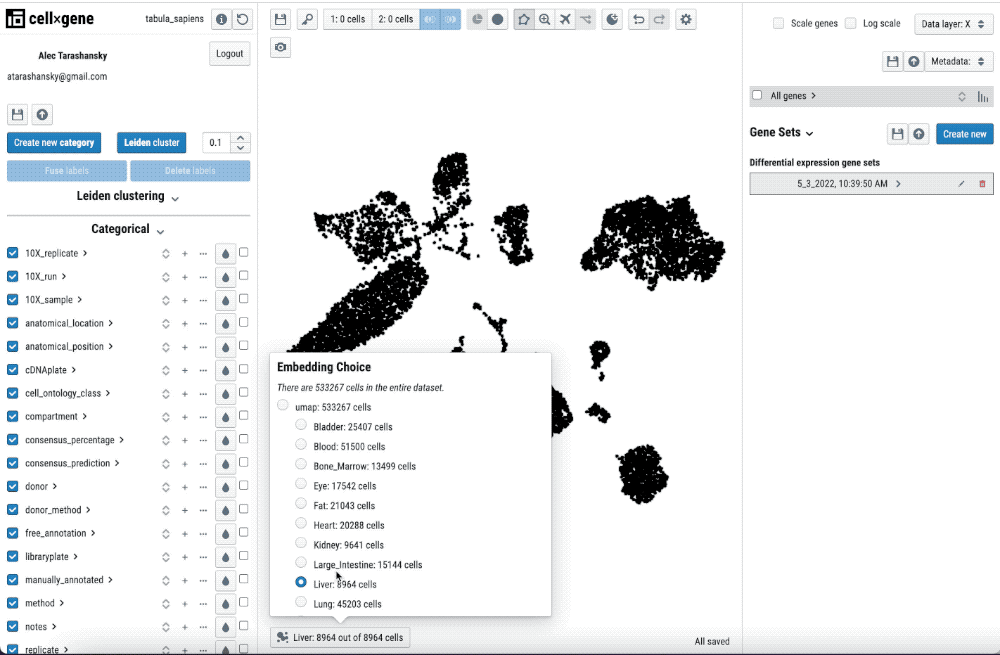
Single-cell transcriptomics analysis requires iteration on pre-processing and annotation of cell-types. Thus, being able to quickly check the pre-processing and its impact in the cell-type annotation and features such as differential gene expression is key to streamline this iterative process.
Tools & Resources
Single-cell transcriptomics analysis requires iteration on pre-processing and annotation of cell-types. Thus, being able to quickly check the pre-processing and its impact in the cell-type annotation and features such as differential gene expression is key to streamline this iterative process.
The Computational Biology team develops open-source tools, datasets, and resources to enable scientific discovery and measure human biology.
protoSpaceJam
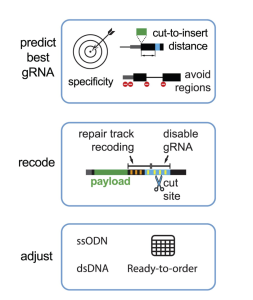 CRISPR/Cas-mediated knock-in of DNA sequences enables precise genome engineering for research and therapeutic applications. However, designing effective guide RNAs (gRNAs) and homology-directed repair (HDR) donors remains a bottleneck.
CRISPR/Cas-mediated knock-in of DNA sequences enables precise genome engineering for research and therapeutic applications. However, designing effective guide RNAs (gRNAs) and homology-directed repair (HDR) donors remains a bottleneck.
Here, we present protoSpaceJAM, an open-source algorithm to automate and optimize gRNA and HDR donor design for CRISPR/Cas insertional knock-in experiments at the genome-wide scale. ProtoSpaceJAM utilizes biological rules to rank gRNAs based on specificity, distance to insertion site, and position relative to regulatory regions. protoSpaceJAM can introduce “recoding” mutations (silent mutations and mutations in non-coding sequences) in HDR donors to prevent re-cutting and increase knock-in efficiency.
Organelle Profiling
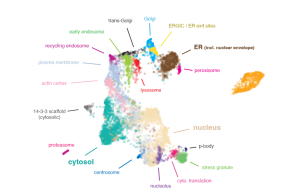 Here, we present a high-resolution strategy to map subcellular organization using organelle immuno-capture coupled to mass spectrometry. A graph-based representation of our data reveals the subcellular localization of over 7,600 proteins, defines spatial protein networks, and uncovers interconnections between cellular compartments. Applying this strategy to characterize the cellular landscape following hCoV-OC43 viral infection, we discover that many proteins are regulated by changes in their spatial distribution rather than by changes in their total abundance.
Here, we present a high-resolution strategy to map subcellular organization using organelle immuno-capture coupled to mass spectrometry. A graph-based representation of our data reveals the subcellular localization of over 7,600 proteins, defines spatial protein networks, and uncovers interconnections between cellular compartments. Applying this strategy to characterize the cellular landscape following hCoV-OC43 viral infection, we discover that many proteins are regulated by changes in their spatial distribution rather than by changes in their total abundance.
Zebrahub
![]() Zebrahub is a multimodal, single-cell RNA sequencing atlas of vertebrate development at single-embryo resolution, using zebrafish as a model organism. The first Zebrahub dataset, of approximately 120,000 cells, spans 10 developmental stages: from end-of-gastrulation embryos to 10-day larvae (bud-stage, 5-, 10-, 15-, 20-, 30-somites stages, as well as 2-, 3-, 5- and 10-days post-fertilization). Four embryos were sequenced per time point. We strive to achieve the highest possible quality; in that context, we expect the Zebrahub dataset to evolve to include more stages and higher data resolution. We collaborate with Biohub’s Royer Group and Genomics Platform on this work.
Zebrahub is a multimodal, single-cell RNA sequencing atlas of vertebrate development at single-embryo resolution, using zebrafish as a model organism. The first Zebrahub dataset, of approximately 120,000 cells, spans 10 developmental stages: from end-of-gastrulation embryos to 10-day larvae (bud-stage, 5-, 10-, 15-, 20-, 30-somites stages, as well as 2-, 3-, 5- and 10-days post-fertilization). Four embryos were sequenced per time point. We strive to achieve the highest possible quality; in that context, we expect the Zebrahub dataset to evolve to include more stages and higher data resolution. We collaborate with Biohub’s Royer Group and Genomics Platform on this work.
AIRRscape
 Technological advances in next-generation sequencing have allowed for broad experimental sampling of immune repertoires, providing insight into how our immune system responds to infection, vaccination, autoimmunity, and cancer. However, the scale of this data can make it difficult to bioinformatically extract the key sequence features that are shared across multiple repertoires.
Technological advances in next-generation sequencing have allowed for broad experimental sampling of immune repertoires, providing insight into how our immune system responds to infection, vaccination, autoimmunity, and cancer. However, the scale of this data can make it difficult to bioinformatically extract the key sequence features that are shared across multiple repertoires.
So we built AIRRscape, an open-source, R Shiny tool to interactively visualize and analyze antibody repertoires. AIRRscape permits users to import and visualize their own AIRR-compliant repertoire data or visualize pre-loaded datasets on any web browser. Get the code on Github.
COVID Tissue Atlas
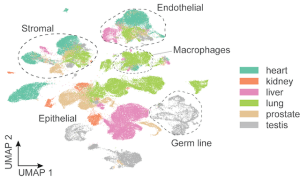 COVID-19 is the most devastating infectious disease in recent history. The pandemic has impacted all parts of the globe and resulted in over 6 million deaths. The systemic effects of severe COVID-19 are largely mediated through the immune response to SARS-CoV-2 infection and subsequent inflammatory response. A multi-organ approach is necessary to improve our understanding of the cellular and molecular mechanisms that drive severe COVID-19 and lead to damage to different organs and tissues.
COVID-19 is the most devastating infectious disease in recent history. The pandemic has impacted all parts of the globe and resulted in over 6 million deaths. The systemic effects of severe COVID-19 are largely mediated through the immune response to SARS-CoV-2 infection and subsequent inflammatory response. A multi-organ approach is necessary to improve our understanding of the cellular and molecular mechanisms that drive severe COVID-19 and lead to damage to different organs and tissues.
We used single-cell transcriptomics to analyze six organs from 15 COVID-positive and five healthy autopsies. Remarkably, through a multi-organ analysis of differential expression and pathway enrichment, we found common transcriptional responses in endothelial cells and macrophages across multiple organs from COVID autopsies. We also identified potential ligand-receptor interactions between these two cell types as targets of signal transduction mechanisms in COVID-19. More generally, our computational efforts provide a basis for analyzing the responses of individual cells while considering the global context of the human body.
exCellxGene
 Single-cell transcriptomics analysis requires iteration on pre-processing and annotation of cell types. Thus, being able to quickly check the pre-processing and its impact on the cell-type annotation and features such as differential gene expression is key to streamlining this iterative process.
Single-cell transcriptomics analysis requires iteration on pre-processing and annotation of cell types. Thus, being able to quickly check the pre-processing and its impact on the cell-type annotation and features such as differential gene expression is key to streamlining this iterative process.
We built Exploratory CellxGene(or exCellxGene) as an extension of CZ CellxGene to assist researchers from the beginning of single-cell omics data analysis (from pre-processing and filtering of data to visualization and annotation) all the way to fast differential gene expression computation.
Napari plug-ins
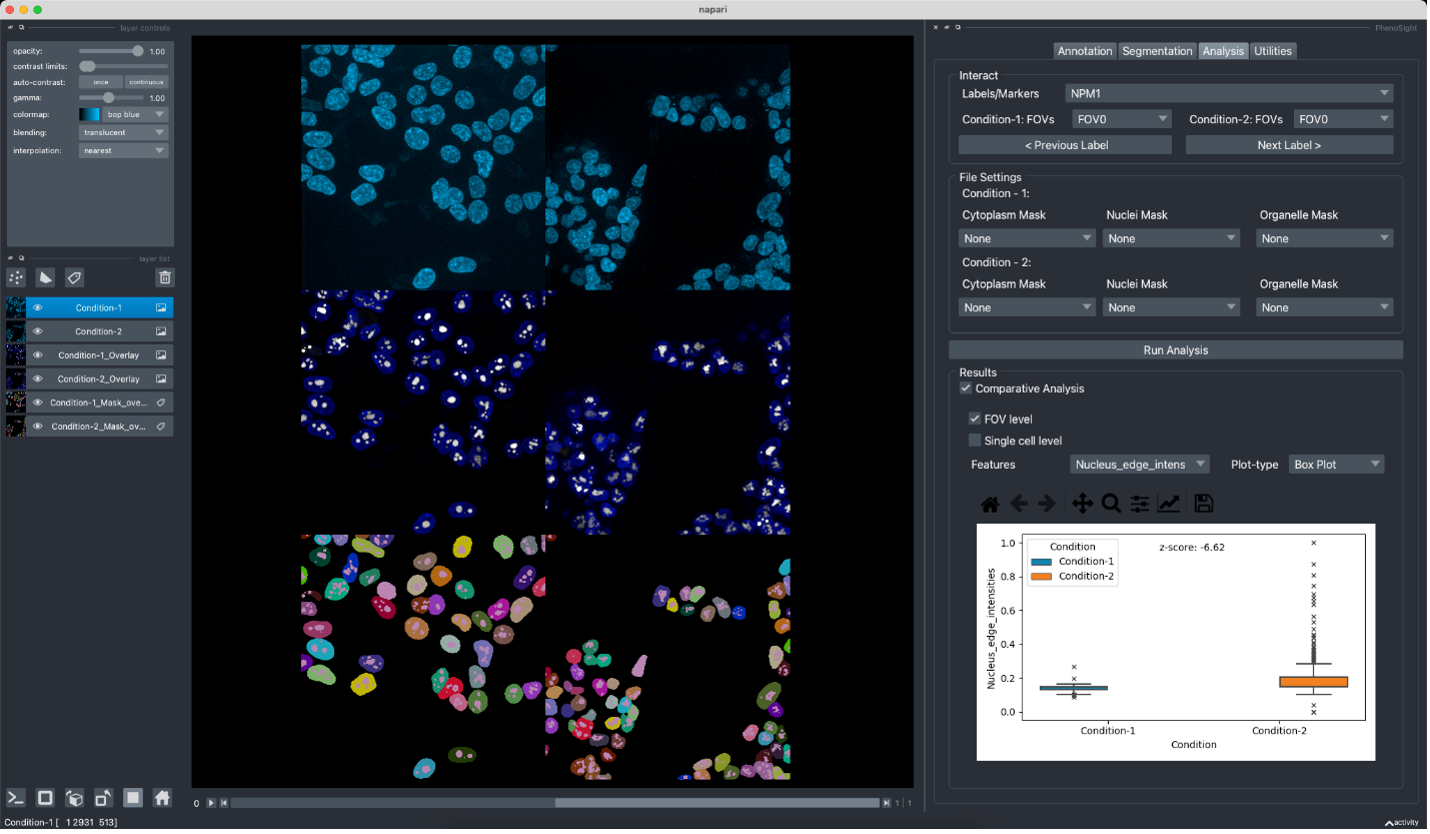 Napari is an easy-to-use open-source image visualization tool for complex scientific datasets.
Napari is an easy-to-use open-source image visualization tool for complex scientific datasets.
We have built napari plug-ins that annotate malaria-infected cells, perform polygonal cropping on multi-scale images, visualize large multimodal MERFISH (Multiplexed Error-Robust Fluorescence In Situ Hybridization) datasets, and interactively perform annotation and comparative phenotyping of perturbed cells.
Ortho_seqs
 ortho_seqs is a Python software tool that quantifies higher order sequence-phenotype interactions based on the previously published multivariate tensor-based orthogonal polynomial method applied to biological sequences.
ortho_seqs is a Python software tool that quantifies higher order sequence-phenotype interactions based on the previously published multivariate tensor-based orthogonal polynomial method applied to biological sequences.
The tool is a packaged command-line utility, installable via PyPI or through Github, and accompanied by an easy-to-use graphical user interface (GUI) along with extensive documentation to allow community use to explore sequence-phenotype relationships.
Spatial Transcriptomics
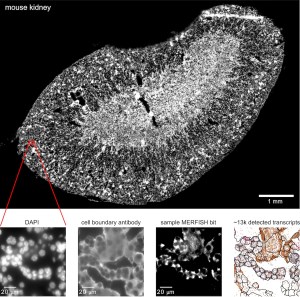 Spatial transcriptomics extends single cell RNA sequencing (scRNA-seq) by providing spatial context for cell type identification and analysis. Imaging-based spatial technologies such as Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH) can achieve unprecedented spatial resolution for applications, such as directly mapping single-cell identities to position within a tissue.
Spatial transcriptomics extends single cell RNA sequencing (scRNA-seq) by providing spatial context for cell type identification and analysis. Imaging-based spatial technologies such as Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH) can achieve unprecedented spatial resolution for applications, such as directly mapping single-cell identities to position within a tissue.
At CZ Biohub, we are utilizing the Vizgen MERSCOPE system to conduct single-molecule spatial transcriptomics for investigating fundamental cell biology, disease states, and embryonic development. By measuring RNA count statistics for hundreds of genes, we can accurately identify cell types within their native spatial context and conduct next-generation bioinformatic analysis.
Tabula Sapiens
Tabula Sapiens is a single-cell transcriptomic atlas of nearly 500,000 cells from 24 different tissues and organs across 14 human donors. This resource provides a rich molecular characterization of more than 800 cell types, their distribution across tissues, and detailed information about tissue-specific variation in gene expression, representing the first draft of a broadly useful reference tool to understand human biology.
Tabula Muris
Tabula Muris is a compendium of single-cell transcriptome data from the mouse derived from nearly 100,000 cells from 20 organs and tissues. The data allow for direct and controlled comparison of gene expression in cell types shared between tissues, such as immune cells from distinct anatomical locations.
Tabula Muris Senis
Tabula Muris Senis provides a rich resource to define the molecular signatures of the aging process in the mouse. It includes data from diverse cell types in 23 tissues and organs across the lifespan, and documents cell-specific changes that occur across multiple cell types and organs, as well as age-related changes in the cellular composition of different organs.
Tabula Microcebus
Tabula Microcebus comprises data from more than 220,000 single cells representing more than 750 defined cell types from 27 organs of the mouse lemur (Microcebus murinus). The atlas provides a cellular and molecular foundation for studying this primate model organism, and a general approach for establishing other emerging model organisms.
Fly Cell Atlas
The Tabula Drosophilae, or Fly Cell Atlas project, sequenced 580,000 cells, annotated by 42 international labs, and provides a molecular characterization of more than 250 cell types covering all tissues of the adult fruit fly, Drosophila melanogaster.

