
By the end of this video, you should understand how we build genetic divergence phylogenetic trees from consensus genomes.
Take-home messages
- When first generated, consensus genomes from different samples will be of different lengths and will have different numbers of sites where the precise nucleotide is known.
- To make sure that we are comparing the same sites in the genome across all of the sequences in our data set, we must perform a multiple sequence alignment.
- When your sequences are all aligned, you can start to see patterns of shared and unique genetic changes. This pattern of shared genetic changes and unique mutations within a sequence is the information that we will encode in the phylogenetic tree.
- We build phylogenetic trees by hierarchically clustering sequences according to which mutations they share. When a mutation occurs on a branch, all the samples that descend from that branch will carry that mutation.
- Mutations that occur earlier in the tree are inherited by more samples.
- Mutations that occur on a terminal branch, or a branch leading to only one sample, are unique to that genome sequence.
Questions
- Given the tree in the figure below, which mutation is shared by genomes A, B, and C? Which mutation is unique to genome C?
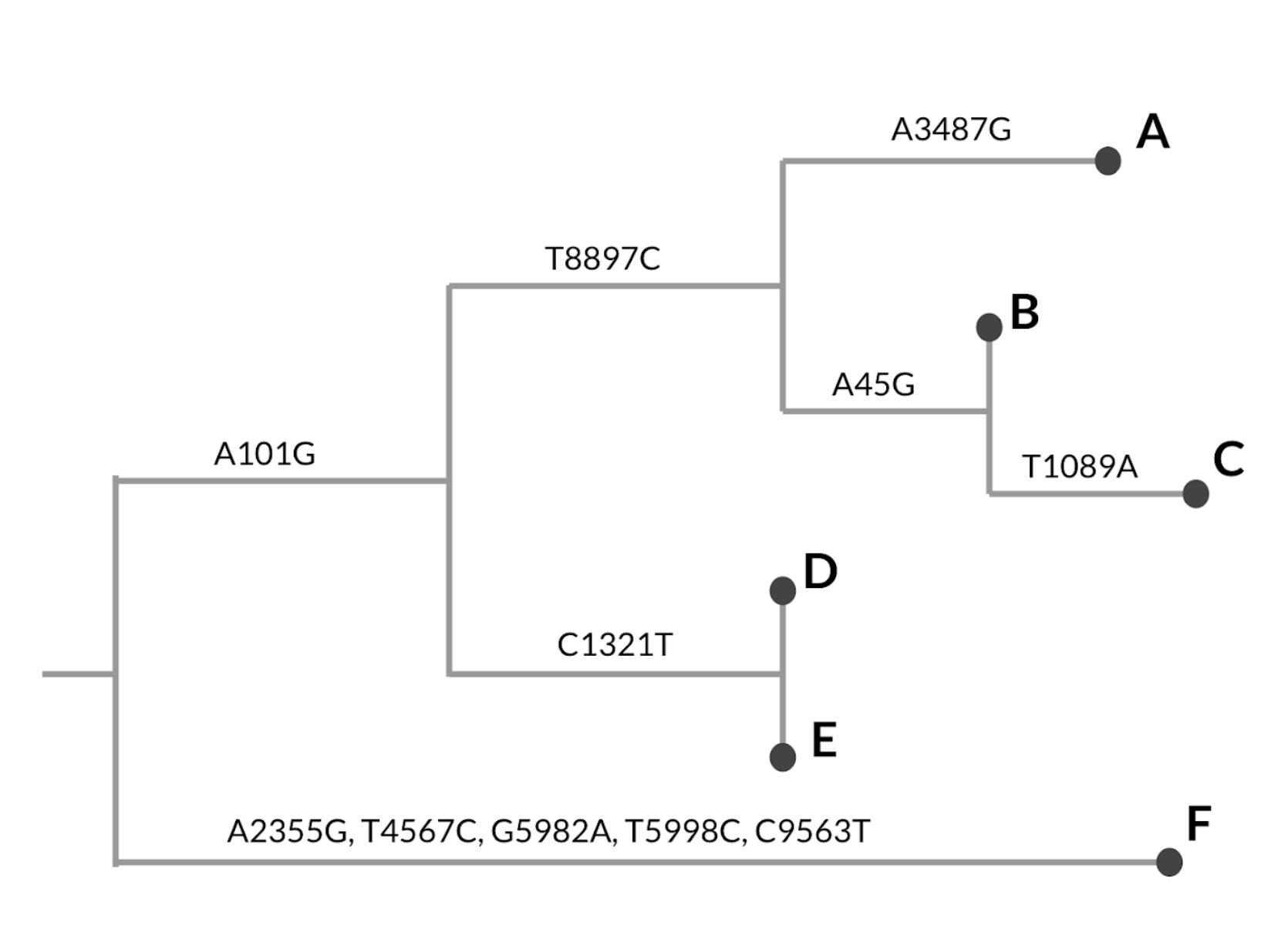
- As we discussed in the video, trees assume that evolution occurs clonally, that is, in a linear fashion in which mutations accrue across a genomic “background”. Given this principle, as well as the patterns of shared mutations that we can see in the tree above, which mutation occurred first: A101G or T8897C?

